Omics Data Collection & Preparation
Overview
Teaching: 30 min
Exercises: 30 minQuestions
What is the experimental design for the data set we are using?
How do I naviagate the terminal?
How should I prepare data for analysis?
What are the most common file formats of transcriptomic and genomic data?
What are some databases and software tools I can use to collect omics data?
Objectives
Gain an understanding of the source of our data.
Become familiar with how transcriptomic and genomic data is structured.
Be able to install and use necessary bioinformatics software tools.
Become familiar with best practices in preparing data for analysis.
Gain hands-on experience and develop skills to be able to use BASH to gather omic data for analysis.
Develop BASH scripting skills to efficiently integrate R scripts into a coherent data analysis pipeline.
Study Design & Data Collection
As a first step, we will begin with collecting some data for analysis. In this workshop we will be using data from a study of the effects of ultraviolet (UV) radiation on the larvae of the red flour beetle, titled “Digital gene expression profiling in larvae of Tribolium castaneum at different periods post UV-B exposure”.

UV radiation is common to many envrioments and it varies in widely in its intensity and composition, such as differing ratios of UV-A and UV-B radiation. The different forms of UV radaition have distinct, and frequently harmful effects on organisms and biological systems. For example, the following diagram depicts the effects of different forms of UV radiation on the skin.
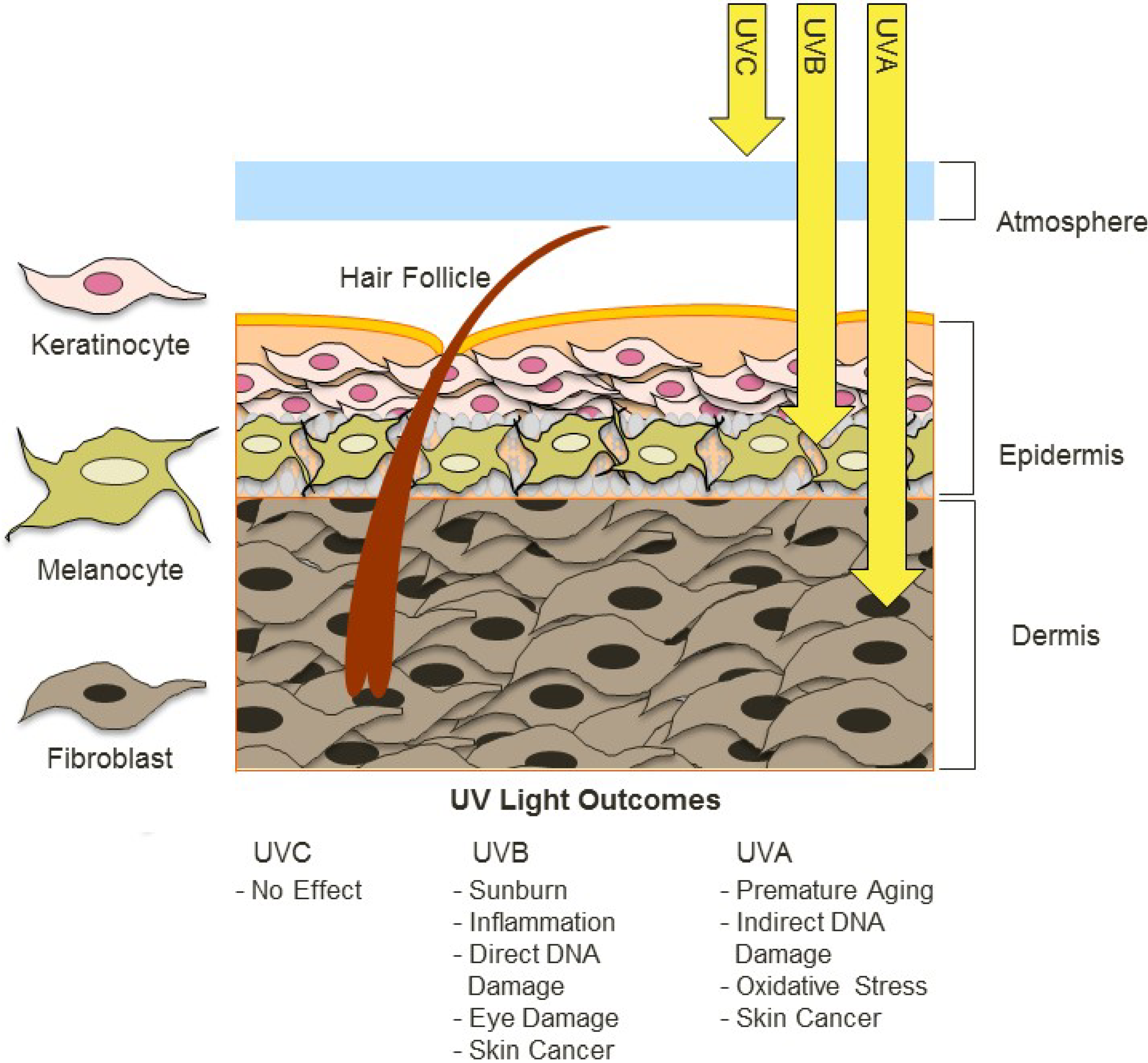
UV radiation is considered an important environmental stressor that organisms need to defend against. There are three primary methods for defending against UV radiation:
- Avoidance
- Photoprotection
- Repair
Since the red flour beetle (Tribolium castaneum) spends much of its life cycle in infested grains, the larvae does not typically experience high levels of UVR. Furthermore, the larvae of the red flour beetle is light in pigmentation and does not appear to employ photoprotective pigments (e.g., melanin) to defend against harmful levels of UV radiation (UVR).
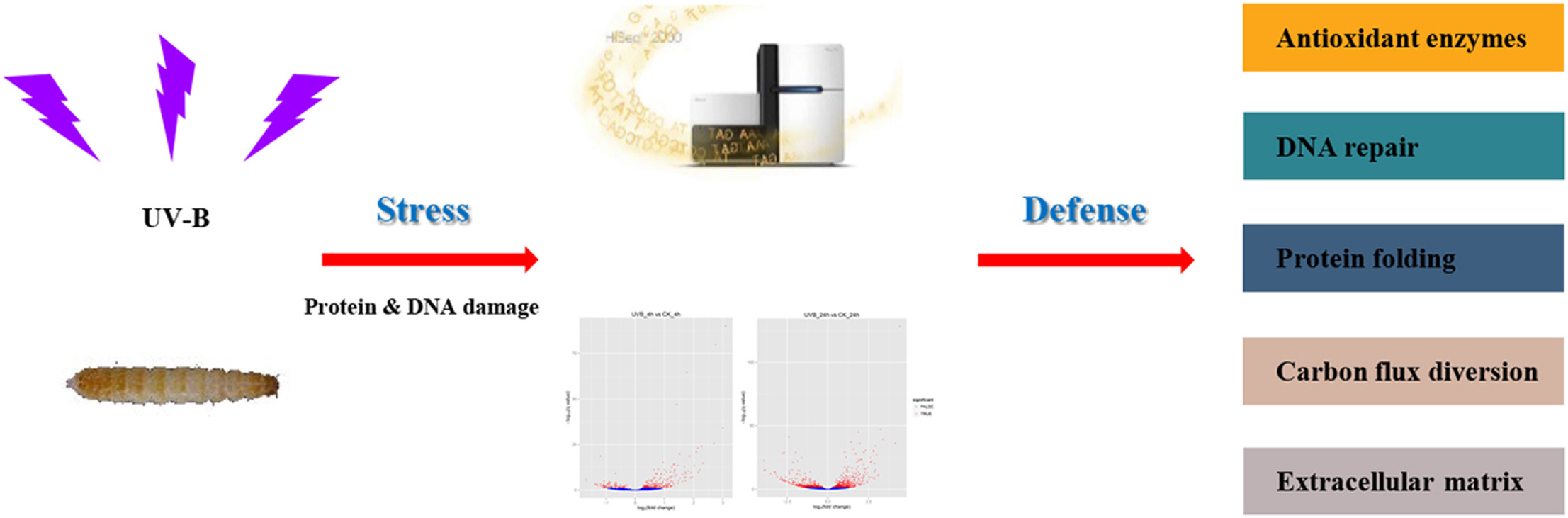
So, how do the larvae handle any exposure to UV radiation? In their study, the authors investigate the defense strategy against UV-B radiation in the red flour beetle. The following graphical abstract illustrates the design of their study, and how the omics data that we will be using in this workshop was collected.
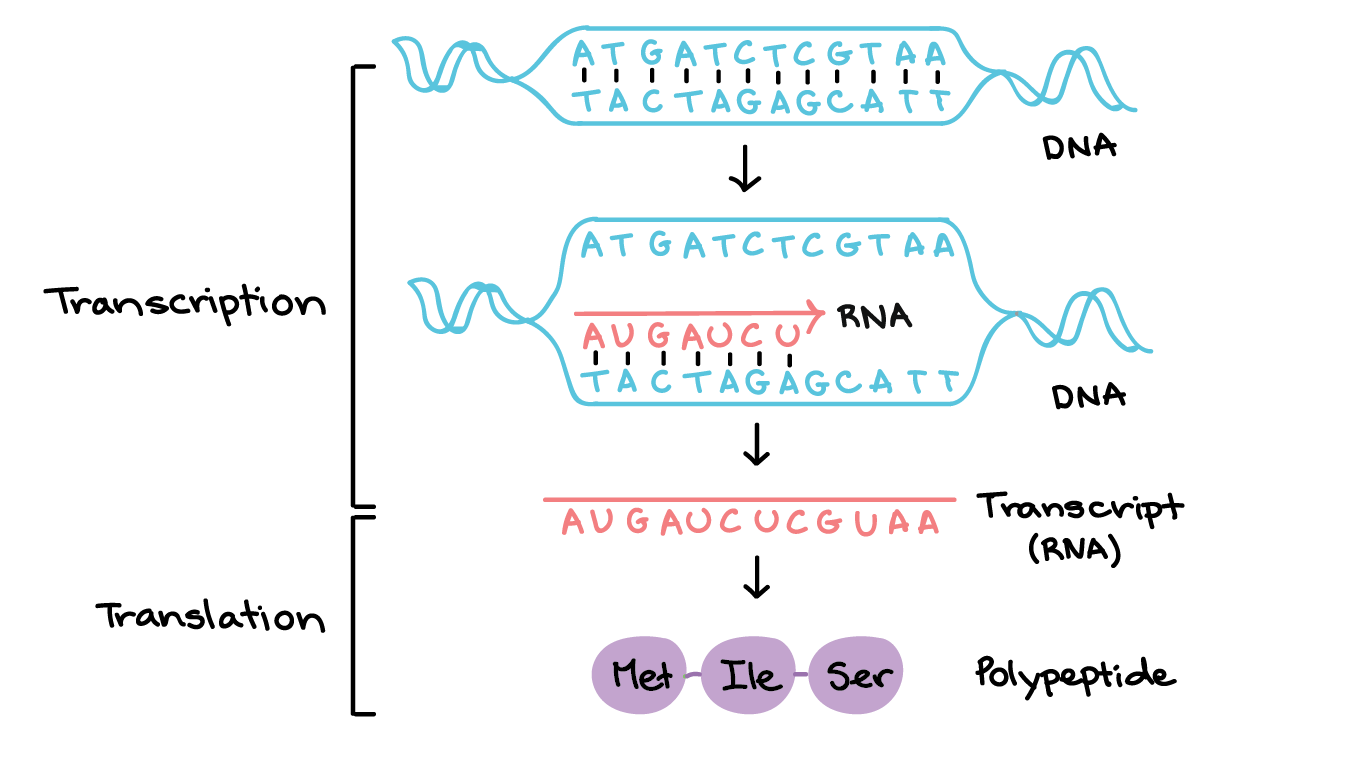
What is Gene Transcription?
Before we proceed with our bioinformatics analysis we should consider the data that we will be working with, and how it is collected. In this workshop we will be using several different file types for the various biological data that we will need in the bioinformatics analysis workflow. Remember that omics technologies include genetic, transcriptomic, proteomic, and metabolomic data.
First, we will need to use transcription sequence data. Transcription is the first step in gene expression, which involves copying the DNA sequence of a gene to make a RNA molecule. For a protein-coding gene, the RNA copy (transcript) carries the information needed to build a polypeptide (protein or protein subunit).
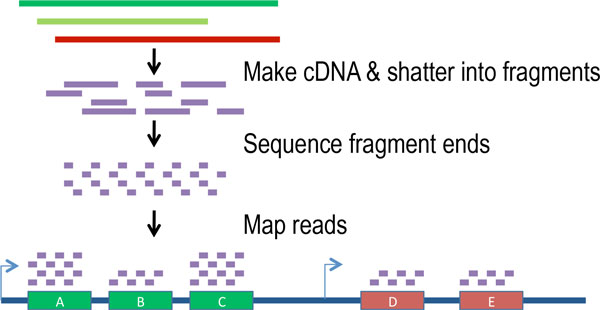
The transcription of genes can be measured using next-generation sequencing techniques that produces millions of sequences (reads) in a short time. This process depicted in the following schematic representation of a RNA sequencing protocol.
Transcription can provide whole genome-wide RNA expression profiles, and is useful for identifying key factors influencing transcription in different environmental conditions. For example, consider the following plot of transcription sequence coverage for a gene model (green) in a species of Daphnia, which has been subjected to a control of visible light (blue) and treatment of UV radiation (red).
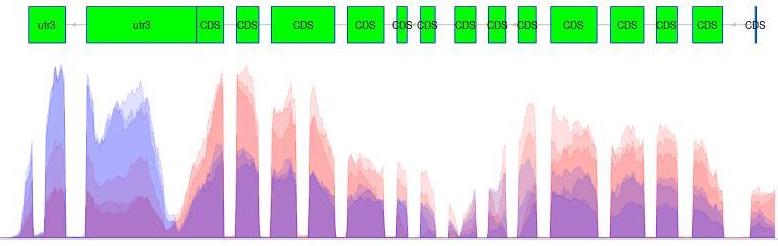
As we can see, the coding sequences (CDS) of the predicted gene model are more highlighy expressed in the tratment of UV radiation. This is visible by the red peaks of transcription sequence read coverage, which are showing the higher frequency of sequence reads from the UV treatment matching up (mapping) to the CDS than the visible light sequences.
Transcriptomic Data Collection
There are several pieces of transcriptomic data that we will need to collect before we can proceed with the bioinformatics analysis. Transcription data are essentially strings of characters in files with very specific formatting.
In this workshop we will be using transcription sequence data in the FASTQ format. This format stores both biological sequences and their corresponding quality scores. Each sequence typically has the following four lines of information.
- A line beginning with @ followed by a sequence identifier and optional description (comment)
- The raw sequence letters
- A line beginning with +, sometimes followed by the same comment as the first line
- A line encoding the quality values for the sequence in line 2, with the same numbers of symbols as letters in the sequence

Software Prerequisite
Note: Make sure that the SRA Toolkit is installed before we proceed to download the transcriptomic data we need. To download the SRA toolkit:
- navigate to the installation instructions
- find the appropriate instructions for your operating system (OS)
Further information and tips for installing the SRA toolkit may be found on the Setup page.
The SRA Toolkit allows you to retrieve data from the SRA for a specific research project using the associated accession number. Additionally, it allows you to produce data file formats like FASTQ using fastq-dump, which is one of the command line tools packaged in the SRA Toolkit.
With the SRA Toolkit installed, we can proceed with collecting the transcriptomic data we need for our bioinformatics analysis. Remeber that we are following the example of the study performed by the authors of “Digital gene expression profiling in larvae of Tribolium castaneum at different periods post UV-B exposure”.
Coding Challenge - Transcriptomic Data Collection
Scripting & Pipelining Tip!
The primary benefit of scripting is the ability to save modular pieces of code to re-use later. As a first step to designing a pipeline, it is a good idea to work through a simple example using data. While you are working through the exercises in this workshop, make sure to store the pieces of code that you write to perform each task.
Recall that the file extension for BASH scripts is .sh.
Note: it is highly recommended that you also write a comment for each piece of code that states why you are using that piece of code. Also, you may wish to leave a comment about the purpose, or what that piece of code does.
Let’s find the transcriptomic data we need by navigating the internet. This data may be accessed by:
Step 1
Go to the paper on the publisher’s website, and be sure you have access to the full article. Recall, “Digital gene expression profiling in larvae of Tribolium castaneum at different periods post UV-B exposure”.
Tip! - Task 1
A simple way to gain access to a specific publication is by searching the name of a paper using the Hesburgh Library website
Step 2
Search the paper for the SRA “accession” number associated with the study.
Tip! - Task 2
Search the paper for “accession” (Mac: command+f, Windows: cntrl+f) and copy the Accession No.
Solution - Task 2
The Accession No. is PRJNA504739 for the transcriptomic data.
Step 3
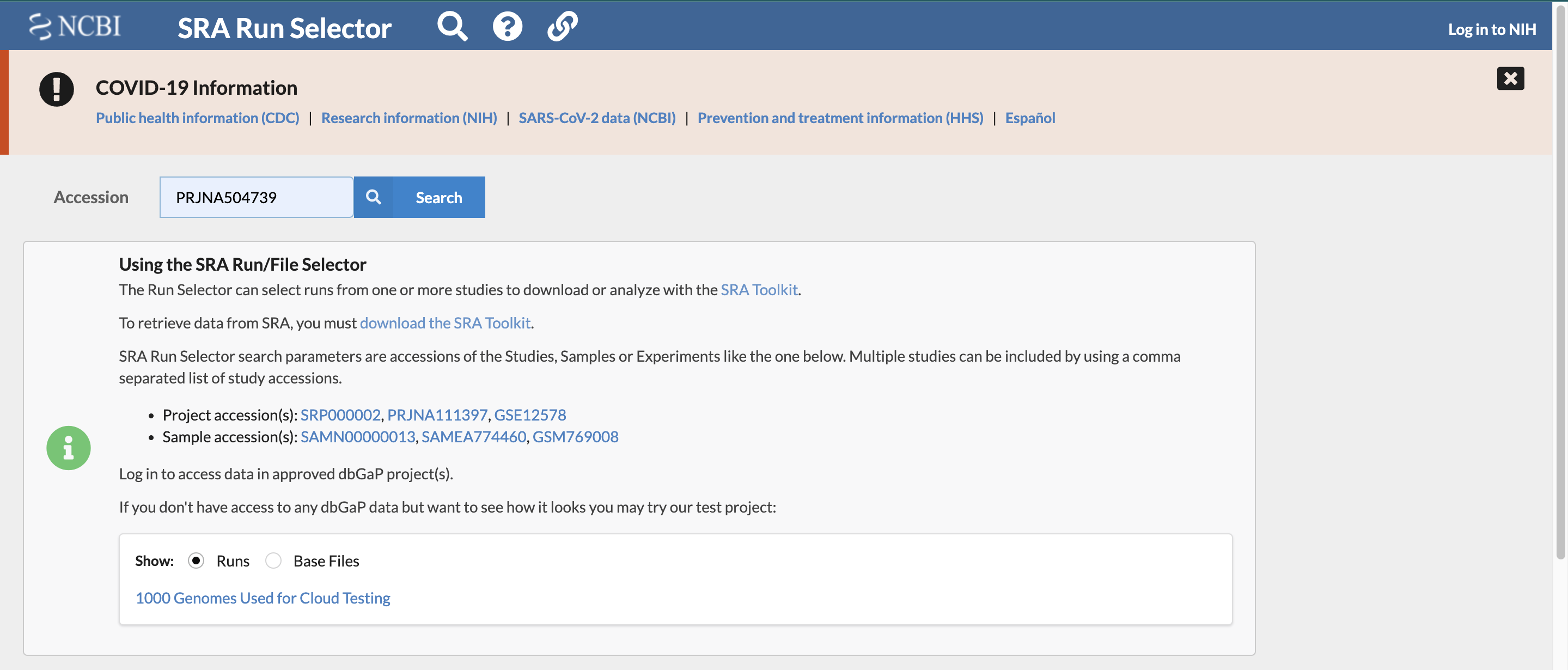
Find the list of accession numbers for the set of transcriptomic data associated with the study by searching for the previously found study accession number using the “SRA Run Selector” on the NCBI website. It is here you will find futher information about each of the sample files as well.
Solution - Task 3
To find the list of accession numbers for the transcripts:
- enter the accession number you found in the paper in the search box as follows:
Step 4
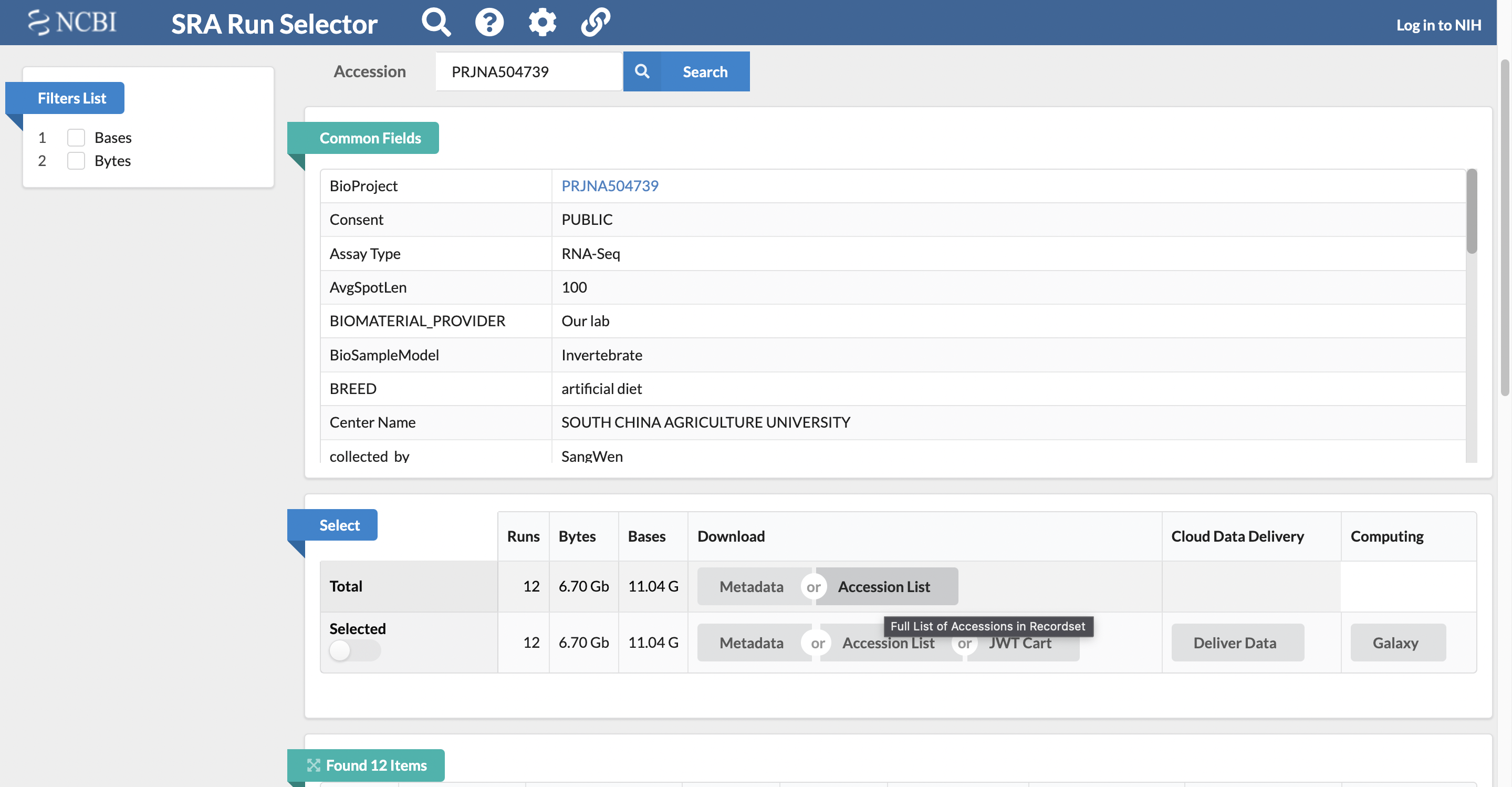
Retrieve the total accession list for the study, which has the SRA accession numbers for all of the samples.
Solution - Task 4
It is possible to download the full list of SRA accession numbers for all the samples associated with the study by selecting to download the total accession list from the “Select” section of the page.
Step 5
Retrieve the all of the transcriptomic sequence data for the study using the SRA accession numbers for all of the samples.
Scripting & Pipelining Tip!
Remember to save these pieces of code in files with the .sh extension While you are working through these exercises!
Solution - Task 5
To retrieve the full set of samples, use the following prefetch and fastq-dump commands and accession numbers. This will take several moments.
prefetch SRR8288561 SRR8288562 SRR8288563 SRR8288564 SRR8288557 SRR8288560 SRR8288558 SRR8288559 SRR8288565 SRR8288566 SRR8288567 SRR8288568 fastq-dump --gzip SRR8288561; fastq-dump --gzip SRR8288562; fastq-dump --gzip SRR8288563; fastq-dump --gzip SRR8288564; fastq-dump --gzip SRR8288557; fastq-dump --gzip SRR8288560 fastq-dump --gzip SRR8288558; fastq-dump --gzip SRR8288559; fastq-dump --gzip SRR8288565; fastq-dump --gzip SRR8288566; fastq-dump --gzip SRR8288567; fastq-dump --gzip SRR8288568
Tip!
Note: that if a terminal command is taking a long time to run, you may use the Cntrl + C keyboard shortcut to kill it and force it to stop running.
Transcriptomic Data Quality Control
An important part of any bioinformatics analysis workflow is the assesment and quality control of your data. In this workshop we are using RNA sequencing reads, which may need to be cleaned if they are raw and include extra pieces of unnecessary data (e.g., adapter sequences.
Software Prerequisites
Note: be sure that you have installed the FastQC software program before we proceed with the bioinformatics analysis workflow.
Further information and tips for installing the FastQC software may be found on the Setup page.
To check if the transcriptomic data that we downloaded has been cleaned already, we need to use the FastQC bioinformatics software tool.
Coding Challenge
Let’s use the following fastqc command to view the quality of one of the sequence read data sets.
fastqc SRR8288561.fastq.gz --extractBe aware that you can also use the FastQC application to view the quality of transcript data using a user interface.
Now we can compare the results that we recieved from the analysis of this sample to the docummentation for FastQC, which include a “Good Illumina Data” example and a “Bad Illumina Data” example.
Genomic Data Collection
Next, we need to collect the neccessary genomic data. Where you get your genomic data may depend on several factors. For example, some or all of the data that you need may be available through an online database.
Checklist
These are common online databases for bioinformatics analysis:
- Database of Genomic Structural Variation (dbVar) – insertions, deletions, duplications, inversions, mobile element insertions, translocations, and complex chromosomal rearrangements
- Database of Genotypes and Phenotypes(dbGaP) - developed to archive and distribute the data and results from studies that have investigated the interaction of genotype and phenotype in Humans
- Database of Single Nucleotide Polymorphisms (dbSNP) - multiple small-scale variations that include insertions/deletions, microsatellites, and non-polymorphic variants
- GenBank - the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences
- Gene - integrates nomenclature, Reference Sequences (RefSeqs), maps, pathways, variations, phenotypes, and links to genome-, phenotype-, and locus-specific from a wide range of species
- Gene Expression Omnibus (GEO) - a public functional genomics data repository supporting MIAME-compliant data submissions
- Gene Expression Omnibus Datasets - stores curated gene expression DataSets, as well as original Series and Platform records in the Gene Expression Omnibus (GEO) repository
- Genome Data Viewer (GDV) - a genome browser supporting the exploration and analysis of more than 380 eukaryotic RefSeq genome assemblies
- International Genome Sample Resource (IGSR) - from the 1000 Genomes Project that ran between 2008 and 2015, which created the largest public catalogue of human variation and genotype data
- The Reference Sequence (RefSeq) - a collection that provides a comprehensive, integrated, non-redundant, well-annotated set of sequences, including genomic DNA, transcripts, and proteins
Depedning on the organisms that you are working with, you may need to locate data in more organism specific databases. We are working with data for the Tribolium castaneum (red flour beetle), which is an arthropod in the order Coleoptera and family Tenebrionidae. So, we are also interested in accessing data from InsectBase. Specifically, we will need:
- reference genome
- genomic features
Challenge
Navigate to the Tribolium castaneum organism page of the InsectBase website and under the “Download” section of the page:
- click the red word Genome
- click the blue word GFF3
This will automatically begin downloading each of the necessary genomic data files.
The reference genome is in the FASTA file format, and has the .fa file extension. This format is commonly used when storing DNA sequence data.

The genomic features file is in the GFF3 file format, and has the .gff3 file extension. This format is a tab-delimited text file that describes the information about any feature of a nucleic acid or protein sequence. These features may include anything from coding sequences (CDS), microRNAs, binding domains, open reading frames (ORFs), etc.
Checklist
GFF3 has 9 required fields, though not all are utilized (either blank or a default value of .):
- Sequence ID - a string with the ID of the sequence
- Source - describes the algorithm or the procedure that generated this feature
- Feature Type - describes what the feature is (mRNA, domain, exon, etc.)
- Feature Start - starting position for the feature
- Feature End - ending position for the feature
- Score - typically E-values for sequence similarity and P-values for predictions
- Strand - a single character that indicates the strand of the feature
- Phase - indicates where the feature begins with reference to the reading frame
- Atributes - a semicolon-separated list of tag-value pairs, providing additional information about each feature
Omics Data Preparation - Command Line
Now that we have the necessary transcript and genomic data, we can begin the process of preparing our data for statistical analysis. This will require proceeding through several steps of the bioinformatics workflow. Recall:
- data collection - SRA toolkit
- quality control - fastqc
- data preparation
- convert genomic data format - gffread
- align transcriptomic data - hisat2
- quantify transcript alignments - featureCounts
- basic statistical analysis and visualization (exact tests) - edgeR
- advanced statistical analysis and visualization (generalized linear models) - edgeR
Software Prerequisites
Note: be sure that you have installed the following software before we proceed with the bioinformatics analysis workflow.
Further information and tips for installing the fastqc, gffread, and hisat2 software programs may be found on the Setup page.
Convert Genomic Data Format
The first step in our bioinformatics workflow is to convert one of the genomic data files to the format expected by our downstread analysis tools. The genome features file that we downloaded from InsectBase for Tribolium castaneum is in the gff3 format, but it needs to be in gtf format to use with Hisat2. This is because there are some important formatting differences between the two genomic feature file types.
Coding Challenge
Let’s use the gffread command line tool to convert the genomic features file from gff3 to gtf format. Hint: check out the manual page for gffread to learn more about the different options.
gffread -E -F -T Tribolium_castaneum.gff3 -o Tribolium.gtf
Align Transcriptomic Data
Next, we need to prepare the transcriptomic sequence data files for statistical analysis by aligning the reads to the reference genome. Recall that our RNA sequence analysis workflow involves mapping the shattered reads to a reference genome.
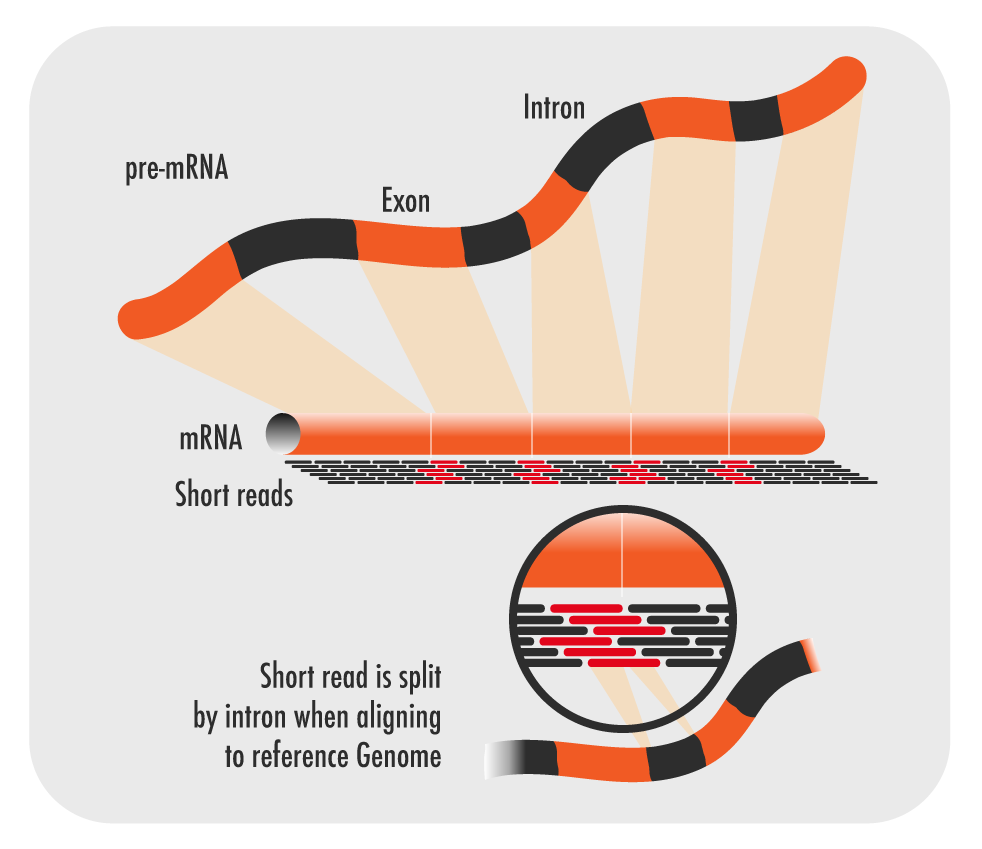
Coding Challenge
Let’s use the Hisat2 command line tool to map the transcriptomic sequence reads to the reference genome of Tribolium castaneum. This is done in two steps. Hint: check out the manual page for Hisat2 to learn more about the different options.
Step 1
First, we need to build a HISAT2 index from the set of DNA sequences for Tribolium castaneum contained in the reference genome fasta file. The build files that are produced by running the following command are needed to map the aligned reads to the reference using the hisat2 commands in the next step (Step 2).
hisat2-build Tribolium_castaneum.genome.fa TriboliumBuildStep 2
Second, we can use the hisat2 command to map each read to the reference genome. This results in an output sam file for each of the input compressed transcript sequence fastq.gz files.
hisat2 -q -x TriboliumBuild -U SRR8288561.fastq.gz -S SRR8288561_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288562.fastq.gz -S SRR8288562_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288563.fastq.gz -S SRR8288563_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288564.fastq.gz -S SRR8288564_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288557.fastq.gz -S SRR8288557_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288560.fastq.gz -S SRR8288560_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288558.fastq.gz -S SRR8288560_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288567.fastq.gz -S SRR8288560_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288568.fastq.gz -S SRR8288560_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288559.fastq.gz -S SRR8288560_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288565.fastq.gz -S SRR8288560_accepted_hits.sam hisat2 -q -x TriboliumBuild -U SRR8288566.fastq.gz -S SRR8288560_accepted_hits.sam
Omics Data Preparation - R
Now that we have the transcript sequence reads aligned to the reference genome, we can begin to quantify the number of reads that map to each genomic feature from the features file. Recall that we are able to count the number of reads that map to each feature in a genome to compare the effects of a treatment, such as UV radiation exposure.
Scripting & Pipelining Tip!
Remember that the primary benefit of scripting is the ability to save modular pieces of code to re-use later. As a first step to designing a pipeline, it is a good idea to work through a simple example using data. While you are working through the exercises in this workshop, make sure to store the pieces of code that you write to perform each task.
After going through your bioinformatics workflow with a simple data example to make an initial scripting pipeline, it is helpful to test your pipeline with a more complicated example. This will allow you to identify the pieces of your pipeline that may easily be generalized for future analysis. For example, defining your script to allow users to enter names for the files they wish to process when they run the script.
Recall that the file extension for R scripts is .r or .R.
Note: it is highly recommended that you also write a comment for each piece of code that states why you are using that piece of code. Also, you may wish to leave a comment about the purpose, or what that piece of code does.
Coding Challenge
Software Prerequisites
Note: be sure that you have loaded the [Rsubread][rsubreadCite] R library before we proceed with the bioinformatics analysis workflow.
Further information and tips for installing the Rsubread R library may be found on the Setup page.
The featureCounts function of the Rsubread library allows us to count the number of transcripts that map to each genomic feature in the Tribolum castaneum reference genome.
Note: the ? symbol can be prepended (e.g., ?featurecounts) to most function names in R to retrieve more information about the function.
Tip!
To make the process easier, remember to set your working directory before proceeding with any coding in R.
setwd("/YOUR/WORKSHOP/DIRECTORY/PATH")Also, remember to load the Rsubread library before you attempt to use the featureCounts function.
library("Rsubread")
As a first step, we need to quantify (count) the number of transcript sequence reads that have mapped (aligned) to the features of the Tribolium castaneum genome.
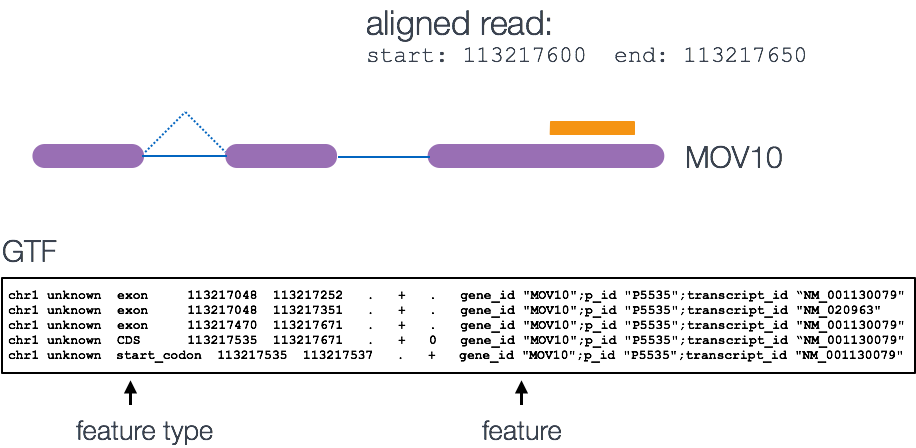
So to quantify the read fragments that map to the genomic features of the red flour beetle genome, we will use the fetureCounts function with the output Hisat2 sam files from the previous step of the bioinformatics workflow.
# control samples at 4h
cntrl1_fc <- featureCounts(files="SRR8288561_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
cntrl2_fc <- featureCounts(files="SRR8288562_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
cntrl3_fc <- featureCounts(files="SRR8288563_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
# control samples 24h
cntrl1_fc_24h <- featureCounts(files="SRR8288558_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
cntrl2_fc_24h <- featureCounts(files="SRR8288567_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
cntrl3_fc_24h <- featureCounts(files="SRR8288568_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
# treatment samples at 4h
treat1_fc <- featureCounts(files="SRR8288564_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
treat2_fc <- featureCounts(files="SRR8288557_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
treat3_fc <- featureCounts(files="SRR8288560_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
# treatment samples 24h
treat1_fc_24h <- featureCounts(files="SRR8288559_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
treat2_fc_24h <- featureCounts(files="SRR8288565_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
treat3_fc_24h <- featureCounts(files="SRR8288566_accepted_hits.sam", annot.ext="Tribolium.gtf", isGTFAnnotationFile=TRUE)
Tip!
Checkout what the resulting data frames looks like using the names and head commands. For example:
names(cntrl1_fc) head(cntrl1_fc$counts)
Before we can move on to any statistical analysis, we need to prepare the data by adding all of the results to a single data frame.
# prepare data frame of sequence read count data
tribolium_counts <- data.frame(
SRR8288561 = unname(cntrl1_fc_4h$counts),
SRR8288562 = unname(cntrl2_fc_4h$counts),
SRR8288563 = unname(cntrl3_fc_4h$counts),
SRR8288558 = unname(cntrl1_fc_24h$counts),
SRR8288567 = unname(cntrl2_fc_24h$counts),
SRR8288568 = unname(cntrl3_fc_24h$counts),
SRR8288564 = unname(treat1_fc_4h$counts),
SRR8288557 = unname(treat2_fc_4h$counts),
SRR8288560 = unname(treat3_fc_4h$counts),
SRR8288559 = unname(treat1_fc_24h$counts),
SRR8288565 = unname(treat2_fc_24h$counts),
SRR8288566 = unname(treat3_fc_24h$counts)
)
# checkout what the resulting counts data frame looks like
head(tribolium_counts)
# set the row names of the counts data frame using a featureCounts results data frame
rownames(tribolium_counts) <- rownames(cntrl1_fc$counts)
# checkout what the updated counts data frame looks like
head(tribolium_counts)
Scripts for this Lesson
Here is the BASH script with the code from this lesson.
Key Points
Explore available omics databases before you design your analysis workflow
Thoroughly explore the supplamental materials of research study papers.
Make sure to install any necessry software in advance.
Always include informative documents for your data.
Carefully name and store your raw and calculated data files.
DE Analysis with Exact Tests
Overview
Teaching: 30 min
Exercises: 30 minQuestions
How do I need to prepare my data for analysis in R?
What standard statistical methods do I need to analyze this data set?
What packages are available to me for biostatistical data analysis in R?
How do I perform basic differential gene expression analysis using R packages?
Objectives
Become familiar with standard statistical methods in R for quantifying transcriptomic gene expression data.
Gain hands-on experience and develop skills to be able to use R to visualize and investigate patterns in omics data.
Be able to install and load the necessary biostatistics R packages.
Be able to run functions from biostatistics packages for data analysis and visualization in R.
Statistical Analysis in R - Exact Tests
With our transcript sequence data now aligned and quantified, we can begin to perform some statistical analysis of the data. For next generation squencing data (e.g., RNA sequences), it is a common task to identify differentially expressed genes (tags) between two (or more) groups. Exact tests often are a good place to start with differential expression analysis of transcriptomic data sets. These tests are the classic edgeR approach to make pairwise comparisons between the groups.
Once negative binomial models are fitted and dispersion estimates are obtained, we can proceed with testing procedures for determining differential expression of the genes in our Tribolium castaneum reference genmoe using the exactTest function of edgeR.
Note: the exact test in edgeR is based on the qCML methods. By knowing the conditional distribution for the sum of counts in a group, we can compute exact p-values by summing over all sums of counts that have a probability less than the probability under the null hypothesis of the observed sum of counts.
Tip!
The exact test for the negative binomial distribution has strong parallels with Fisher’s exact test, and is only applicable to experiments with a single factor. The Fisher’s exact test is used to determine if there is a significant association between two categorical variables. And it is typically used as an alternative to the Chi-Square Test of Independence when the data is small.
So, the types of contrasts you can make will depend on the design of your study and data set. The experimental design of the data we are using in this workshop is as follows:
| sample | treatment | hours |
|---|---|---|
| SRR8288561 | cntrl | 4h |
| SRR8288562 | cntrl | 4h |
| SRR8288563 | cntrl | 4h |
| SRR8288564 | treat | 4h |
| SRR8288557 | treat | 4h |
| SRR8288560 | treat | 4h |
| SRR8288558 | cntrl | 24h |
| SRR8288567 | cntrl | 24h |
| SRR8288568 | cntrl | 24h |
| SRR8288559 | treat | 24h |
| SRR8288565 | treat | 24h |
| SRR8288566 | treat | 24h |
Note in the treatment column:
- cntrl - control treatment
- treat - treatment of UV-B exposure
It is apparent from the above experimental design layout that we are at least able to perform exact tests (t-tests) with the read count (gene expression) data.
After normalization of raw counts we will perform genewise exact tests for differences in the means between two groups of gene counts. Specifically, the two experimental groups of treatment and control for the Tribolium castaneum transcript sequence data.
Software Prerequisites
Note: be sure that you have loaded the edgeR R library before we proceed with the bioinformatics analysis workflow.
Further information and tips for installing the edgeR R library may be found on the Setup page.
We will use the edgeR library in R with the data frame of transcript sequence read counts from the previous step of the bioinformatics workflow.
Tip!
Remember to load the edgeR library before you attempt to use the following functions.
library("edgeR")
Before we proceed with conducting any analysis in R, we should set our working directory to the location of our data. Then, we need to import our data using the read.csv function.
# set the working directory
setwd("/YOUR/FILE/PATH/")
# import gene count data
tribolium_counts <- read.csv("TriboliumCounts.csv", row.names="X")
As a first step in the analysis, we need to describe the layout of samples in our transcript sequence read count data frame using a list object.
# add grouping factor to specify the layout of the count data frame
group <- factor(c(rep("cntrl_4h",3), rep("treat_4h",3), rep("cntrl_24h",3), rep("treat_24h",3)))
# create DGE list object
list <- DGEList(counts=tribolium_counts,group=group)
This is a good point to generate some interesting plots of our input data set before we begin preparing the raw gene counts for the exact test.
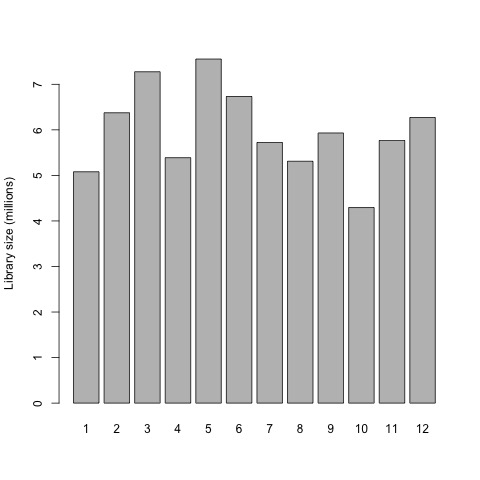
So first, we will now plot the library sizes of our sequencing reads before normalization using the barplot function.
# plot the library sizes before normalization
barplot(list$samples$lib.size*1e-6, names=1:12, ylab="Library size (millions)")
Plot
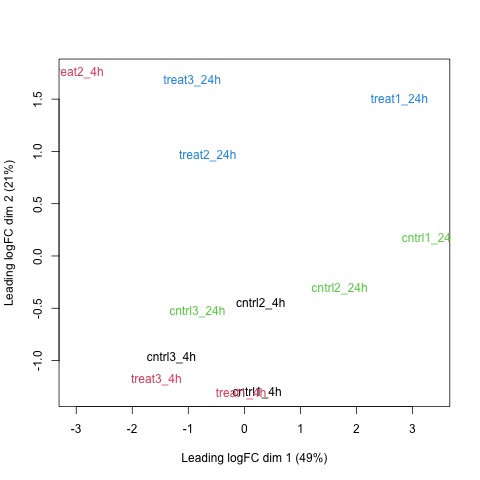
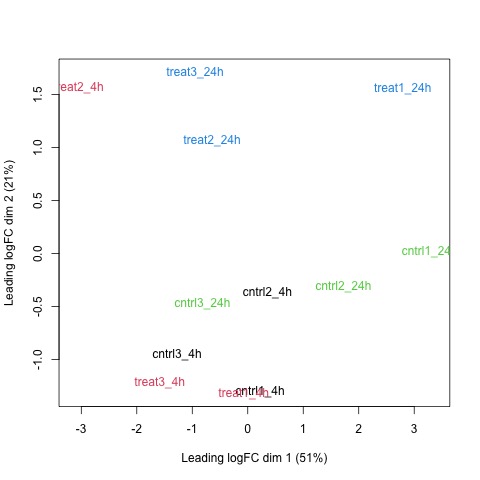
Next, we will use the plotMDS function to display the relative similarities of the samples and view batch and treatment effects before normalization.
# draw a MDS plot to show the relative similarities of the samples
# and to view batch and treatment effects before normalization
plotMDS(list, col=rep(1:12, each=3))
Plot
There is no purpose in analyzing genes that are not expressed in either experimental condition (treatment or control), so raw gene counts are first filtered by expression levels.
# gene expression is first filtered based on expression levels
keep <- filterByExpr(list)
table(keep)
list <- list[keep, , keep.lib.sizes=FALSE]
# calculate normalized factors
list <- calcNormFactors(list)
normList <- cpm(list, normalized.lib.sizes=TRUE)
#Write the normalized counts to a file
write.table(normList, file="tribolium_normalizedCounts.csv", sep=",", row.names=TRUE)
# view normalization factors
list$samples
dim(list)
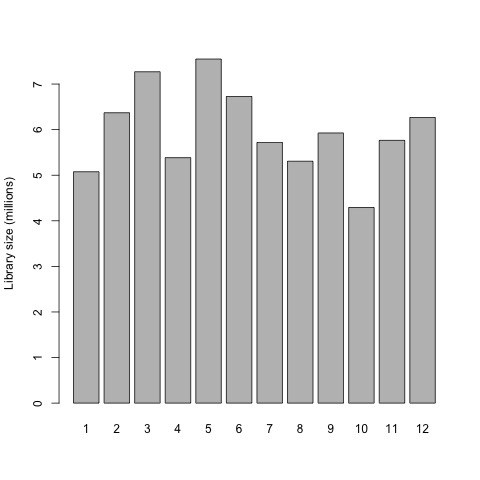
Now that we have normalized gene counts for our samples we should generate the same set of previous plots for comparison.
# plot the library sizes after normalization
barplot(list$samples$lib.size*1e-6, names=1:12, ylab="Library size (millions)")
# draw a MDS plot to show the relative similarities of the samples
# and to view batch and treatment effects after normalization
plotMDS(list, col=rep(1:12, each=3))
Plots
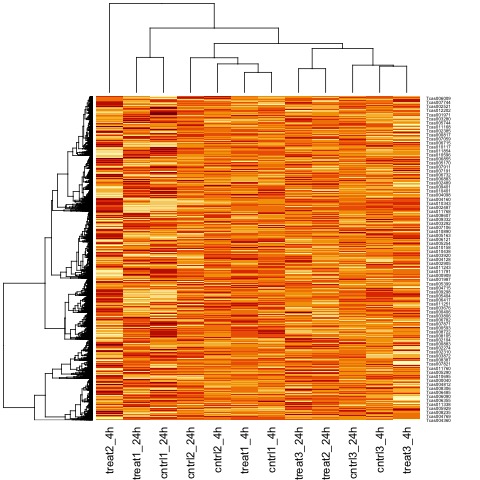
It can also be useful to view the moderated log-counts-per-million after normalization using the cpm function results with heatmap.
# draw a heatmap of individual RNA-seq samples using moderated
# log-counts-per-million after normalization
logcpm <- cpm(list, log=TRUE)
heatmap(logcpm)
Plot
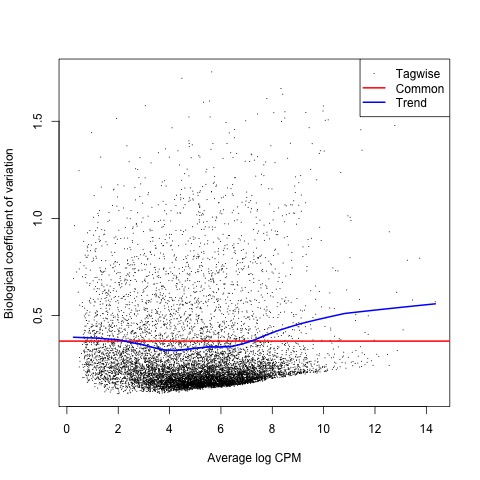
With the normalized gene counts we can also produce a matrix of pseudo-counts to estimate the common and tagwise dispersions. This allows us to use the plotBCV function to generate a genewise biological coefficient of variation (BCV) plot of dispersion estimates.
# produce a matrix of pseudo-counts and
# estimate common dispersion and tagwise dispersions
list <- estimateDisp(list)
list$common.dispersion
# view dispersion estimates and biological coefficient of variation
plotBCV(list)
Plot
Exact Tests - treat_4h vs ctrl_4h
Now, we are ready to perform exact tests with edgeR using the exactTest function.
#Perform an exact test for treat_4h vs ctrl_4h
tested <- exactTest(list, pair=c("cntrl_4h", "treat_4h"))
#Create results table of DE genes
resultsTbl <- topTags(tested, n=nrow(tested$table))$table
#Create filtered results table of DE genes
resultsTbl.keep <- resultsTbl$FDR <= 0.05
resultsTblFiltered <- resultsTbl[resultsTbl.keep,]
Using the resulting differentially expressed (DE) genes from the exact test we can view the counts per million for the top genes of each sample.
# look at the counts-per-million in individual samples for the top genes
o <- order(tested$table$PValue)
cpm(list)[o[1:10],]
# view the total number of differentially expressed genes at a p-value of 0.05
summary(decideTests(tested))
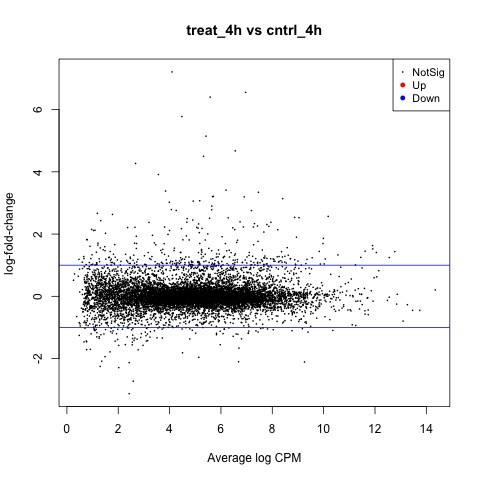
We can also generate a mean difference (MD) plot of the log fold change (logFC) against the log counts per million (logcpm) using the plotMD function. DE genes are highlighted and the blue lines indicate 2-fold changes.
# plot log-fold change against log-counts per million, with DE genes highlighted
# the blue lines indicate 2-fold changes
plotMD(tested)
abline(h=c(-1, 1), col="blue")
Plot
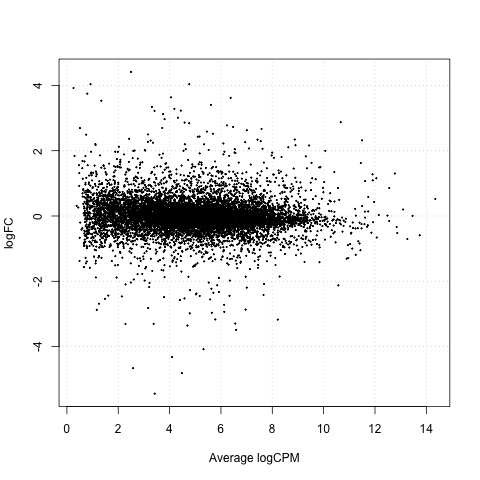
As a final step, we will produce a MA plot of the libraries of count data using the plotSmear function. There are smearing points with very low counts, particularly those counts that are zero for one of the columns.
# make a mean-difference plot of the libraries of count data
plotSmear(tested)
Plot
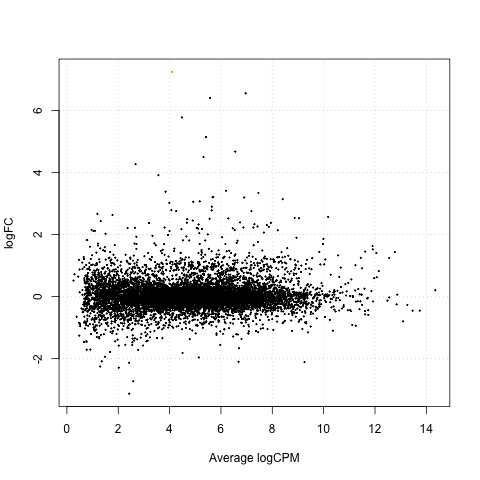
Exact Tests - treat_24h vs ctrl_24h
Next, we will perform exact tests on the 24h data with edgeR using the exactTest function.
#Perform an exact test for treat_24h vs ctrl_24h
tested_24h <- exactTest(list, pair=c("cntrl_24h", "treat_24h"))
#Create a table of DE genes filtered by FDR
resultsTbl_24h <- topTags(tested_24h, n=nrow(tested_24h$table))$table
#Create filtered results table of DE genes
resultsTbl_24h.keep <- resultsTbl_24h$FDR <= 0.05
resultsTbl_24h_filtered <- resultsTbl_24h[resultsTbl_24h.keep,]
#Write the results of the exact tests to a csv file
write.table(resultsTbl_24h_filtered, file="exactTest_24h_filtered.csv", sep=",", row.names=TRUE)
Using the resulting differentially expressed (DE) genes from the exact test we can view the counts per million for the top genes of each sample.
#Look at the counts-per-million in individual samples for the top genes
o <- order(tested_24h$table$PValue)
cpm(list)[o[1:10],]
# view the total number of differentially expressed genes at a p-value of 0.05
summary(decideTests(tested_24h))
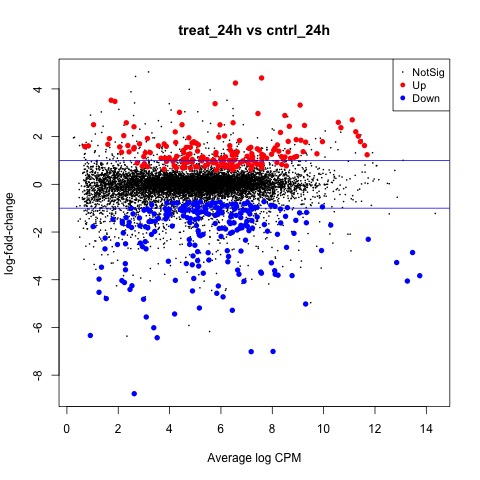
We can also generate a mean difference (MD) plot of the log fold change (logFC) against the log counts per million (logcpm) using the plotMD function. DE genes are highlighted and the blue lines indicate 2-fold changes.
# plot log-fold change against log-counts per million, with DE genes highlighted
# the blue lines indicate 2-fold changes
plotMD(tested_24h)
abline(h=c(-1, 1), col="blue")
Plot
As a final step, we will produce a MA plot of the libraries of count data using the plotSmear function. There are smearing points with very low counts, particularly those counts that are zero for one of the columns.
# make a mean-difference plot of the libraries of count data
plotSmear(tested_24h)
Plot
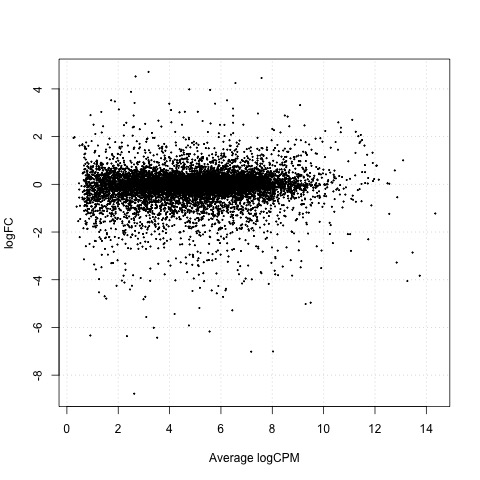
Exact Tests - treat_4h vs treat_24h
Next, we will perform exact tests on the treatment data with edgeR using the exactTest function.
#Perform an exact test for treat_4h vs treat_24h
tested_treat <- exactTest(list, pair=c("treat_24h", "treat_4h"))
#Create a table of DE genes filtered by FDR
resultsTbl_treat <- topTags(tested_treat, n=nrow(tested_treat$table))$table
#Create filtered results table of DE genes
resultsTbl_treat.keep <- resultsTbl_treat$FDR <= 0.05
resultsTbl_treat_filtered <- resultsTbl_treat[resultsTbl_treat.keep,]
#Write the results of the exact tests to a csv file
write.table(resultsTbl_treat_filtered, file="exactTest_treat_filtered.csv", sep=",", row.names=TRUE)
Using the resulting differentially expressed (DE) genes from the exact test we can view the counts per million for the top genes of each sample.
#Look at the counts-per-million in individual samples for the top genes
o <- order(tested_treat$table$PValue)
cpm(list)[o[1:10],]
# view the total number of differentially expressed genes at a p-value of 0.05
summary(decideTests(tested_treat))
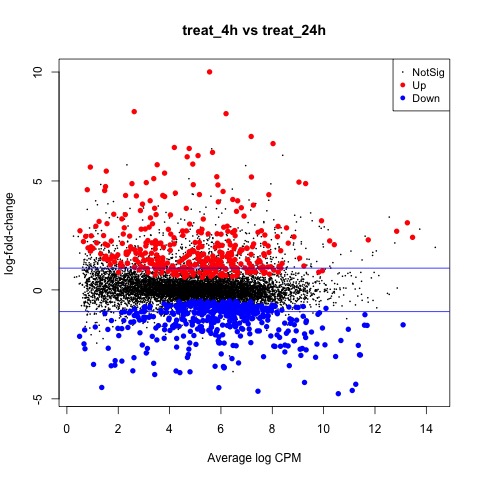
We can also generate a mean difference (MD) plot of the log fold change (logFC) against the log counts per million (logcpm) using the plotMD function. DE genes are highlighted and the blue lines indicate 2-fold changes.
# plot log-fold change against log-counts per million, with DE genes highlighted
# the blue lines indicate 2-fold changes
plotMD(tested_treat)
abline(h=c(-1, 1), col="blue")
Plot
As a final step, we will produce a MA plot of the libraries of count data using the plotSmear function. There are smearing points with very low counts, particularly those counts that are zero for one of the columns.
# make a mean-difference plot of the libraries of count data
plotSmear(tested_treat)
Plot
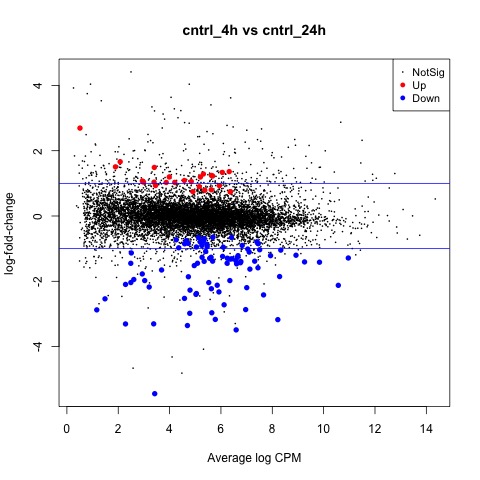
Exact Tests - cntrl_4h vs cntrl_24h
Next, we will perform exact tests on the control data with edgeR using the exactTest function.
#Perform an exact test for cntrl_4h vs ctrl_24h
tested_cntrl <- exactTest(list, pair=c("cntrl_24h", "cntrl_4h"))
#Create a table of DE genes filtered by FDR
resultsTbl_nctrl <- topTags(tested_cntrl, n=nrow(tested_cntrl$table))$table
#Create filtered results table of DE genes
resultsTbl_nctrl.keep <- resultsTbl_nctrl$FDR <= 0.05
resultsTbl_treat_filtered <- resultsTbl_nctrl[resultsTbl_nctrl.keep,]
#Write the results of the exact tests to a csv file
write.table(resultsTbl_cntrl_filtered, file="exactTest_cntrl_filtered.csv", sep=",", row.names=TRUE)
Using the resulting differentially expressed (DE) genes from the exact test we can view the counts per million for the top genes of each sample.
#Look at the counts-per-million in individual samples for the top genes
o <- order(tested_cntrl$table$PValue)
cpm(list)[o[1:10],]
# view the total number of differentially expressed genes at a p-value of 0.05
summary(decideTests(tested_cntrl))
We can also generate a mean difference (MD) plot of the log fold change (logFC) against the log counts per million (logcpm) using the plotMD function. DE genes are highlighted and the blue lines indicate 2-fold changes.
# plot log-fold change against log-counts per million, with DE genes highlighted
# the blue lines indicate 2-fold changes
plotMD(tested_cntrl)
abline(h=c(-1, 1), col="blue")
Plot
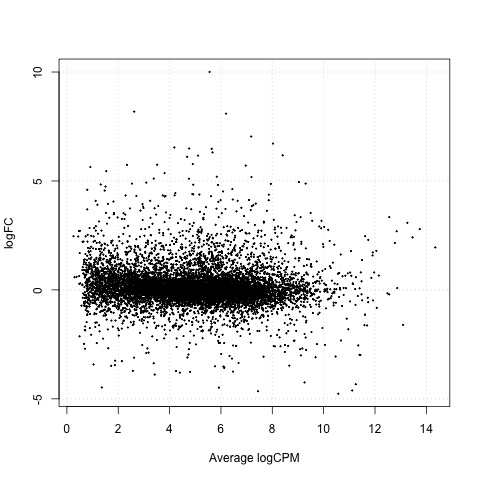
As a final step, we will produce a MA plot of the libraries of count data using the plotSmear function. There are smearing points with very low counts, particularly those counts that are zero for one of the columns.
# make a mean-difference plot of the libraries of count data
plotSmear(tested_cntrl)
Plot
Scripts for this Lesson
Here is the R script with the code from this lesson. Also included is a R script with the plots being written to jpg files, rather than output to the screen.
Key Points
The BiocManager is is a great tool for installing Bioconductor packages in R.
Make sure to use the ? symbol to check the documentation for most R functions.
Fragments of sequences from RNA sequencing may be mapped to a reference genome and quantified.
The edgeR manual has many good examples of differential expression analysis on different data sets.
Break
Overview
Teaching: 0 min
Exercises: 10 minQuestions
Take a break!
Objectives
Take a break!
Key Points
Take a break!
DE Analysis with Generalized Linear Models
Overview
Teaching: 30 min
Exercises: 50 minQuestions
How do I need to prepare my data for analysis in R?
What standard statistical methods do I need to analyze this data set?
What packages are available to me for biostatistical data analysis in R?
How do I perform advanced differential gene expression analysis using R packages?
Objectives
Become familiar with advanced statistical methods in R for quantifying transcriptomic gene expression data.
Gain hands-on experience and develop skills to be able to use R to visualize and investigate patterns in omics data.
Be able to install and load the necessary biostatistics R packages.
Be able to run functions from biostatistics packages for data analysis and visualization in R.
Advanced Statistical Analysis in R - Generalized Linear Models
With our transcript sequence data now aligned and quantified, we can also use generalized linear models (GLM). These are another classic edgeR method for analyzing RNA-seq expression data. In contrast to exact tests, GLMs allow for more general comparisons.
Note: GLMs are an extension of classical linear models to nonnormally distributed response data. GLMs specify probability distributions according to their mean-variance relationship.
Tip!
GLM’s may be used for general experiments with multiple factors, and parallels the ANOVA method. This is in contrast to the exact tests, which are only applicable to experiments with a single factor.
As usual, the types of comparisons you can make will depend on the design of your study. In our case:
| sample | treatment | hours |
|---|---|---|
| SRR8288561 | cntrl | 4h |
| SRR8288562 | cntrl | 4h |
| SRR8288563 | cntrl | 4h |
| SRR8288564 | treat | 4h |
| SRR8288557 | treat | 4h |
| SRR8288560 | treat | 4h |
| SRR8288558 | cntrl | 24h |
| SRR8288567 | cntrl | 24h |
| SRR8288568 | cntrl | 24h |
| SRR8288559 | treat | 24h |
| SRR8288565 | treat | 24h |
| SRR8288566 | treat | 24h |
Note in the treatment column:
- cntrl - control treatment
- treat - treatment of UV-B exposure
So, from the above experimental design layout that we can see that we have the ability to perform an ANOVA like tests using generalized linear models with the read count (gene expression) data.
After normalization of the raw gene counts we will perform genewise quasi-likelihood (QL) F-tests using GLMs in edgeR. These tests allow us to perform ANOVA like tests for differences in the means within and between our grouping factors.
The hypotheses we will be testing:
- the means of the treatment factor are equal
- the means of the hours factor are equal
- there is no interaction between the two factors
Note that, we choose to use QL F-tests over likelihood ratio tests (LRT) since it maintains the uncertainty in estimating the dispersion for each gene.
Tip!
Since we have a more complicated experimental design layout, we will be using a csv file to specify the design.
targets <- read.csv(file="groupingFactors.csv")The groupingFactors.csv file has the following layout.

We will use the edgeR library in R with the data frame of transcript sequence read counts from the previous step of the bioinformatics workflow.
Tip!
Remember to load the edgeR library before you attempt to use the following functions.
library("edgeR")
Before we proceed with conducting any analysis in R, we should set our working directory to the location of our data. Then, we need to import our data using the read.csv function.
# set the working directory
setwd("/YOUR/FILE/PATH/")
# import gene count data
tribolium_counts <- read.csv("TriboliumCounts.csv", row.names="X")
As a first step in the analysis, we need to setup the design matrix describing the experimental design layout of the study.
# setup a design matrix
group <- factor(paste(targets$treatment,targets$hours,sep="."))
The grouping factors need to be added as a column to our experimental design before we can create a DGE list object. Here we use paste and factor to combine the factors (treatment and hours) into one string (group) separated by a period for each sample.
# create DGE list object
list <- DGEList(counts=tribolium_counts,group=group)
colnames(list) <- rownames(targets)
This is a good point to generate some interesting plots of our input data set before we begin preparing the raw gene counts for the exact test. So first, we will now plot the library sizes of our sequencing reads before normalization using the barplot function.
# plot the library sizes before normalization
barplot(list$samples$lib.size*1e-6, names=1:12, ylab="Library size (millions)")
Plot
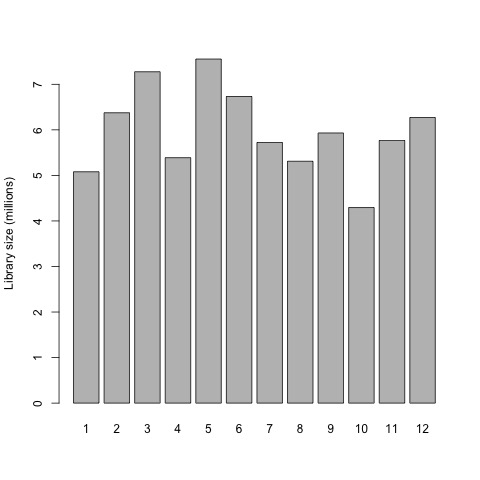
Next, we need to filter the raw gene counts by expression levels and remove counts of lowly expressed genes.
#Retain genes only if it is expressed at a minimum level
keep <- filterByExpr(list)
summary(keep)
list <- list[keep, , keep.lib.sizes=FALSE]
The filtered raw counts are then normalized with calcNormFactors according to the weighted trimmed mean of M-values (TMM) to eliminate composition biases between libraries. The normalized gene counts are output in counts per million (CPM).
#Use TMM normalization to eliminate composition biases
# between libraries
list <- calcNormFactors(list)
#Retrieve normalized counts
normList <- cpm(list, normalized.lib.sizes=TRUE)
#Write the normalized counts to a file
write.table(normList, file="tribolium_normalizedCounts.csv", sep=",", row.names=TRUE)
#View normalization factors
list$samples
dim(list)
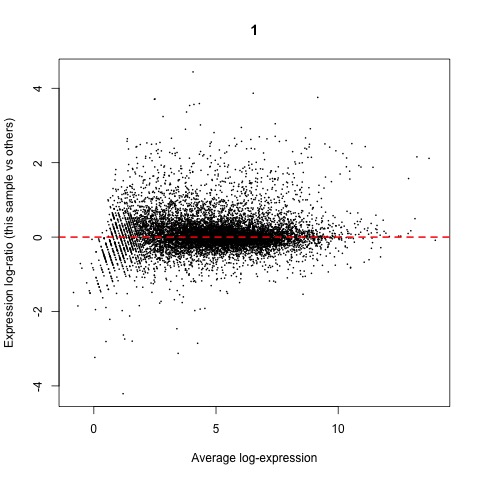
Now that we have normalized gene counts for our samples we should generate some informative plots of our normalized data. First, we can verify the TMM normalization with a mean difference (MD) plot of all log fold change (logFC) against average count size.
#Verify TMM normalization using a MD plot
plotMD(cpm(list, log=TRUE), column=1)
abline(h=0, col="red", lty=2, lwd=2)
Plot
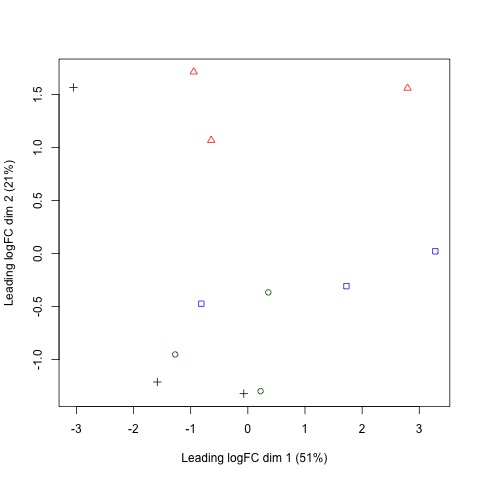
Next, we will use plotMDS to display the relative similarities of the samples and view the differences between the expression profiles of different samples.
Tips!
A few things to keep in mind:
- a legend is added to the top left corner of the plot by specifying “topleft” in the legend function. The location of the legend may be changed by updating the first argument of the function.
- the data frames of the points and colors will need to be updated if you have a different number of sample sets than in our example. These data frames are used to set the pch and col arguments of the plotMDS function.
#Use a MDS plot to visualizes the differences
# between the expression profiles of different samples
points <- c(0,1,2,3,15,16,17,18)
colors <- rep(c("blue", "darkgreen", "red", "black"), 2)
#Create plot without legend
plotMDS(list, col=colors[group], pch=points[group])
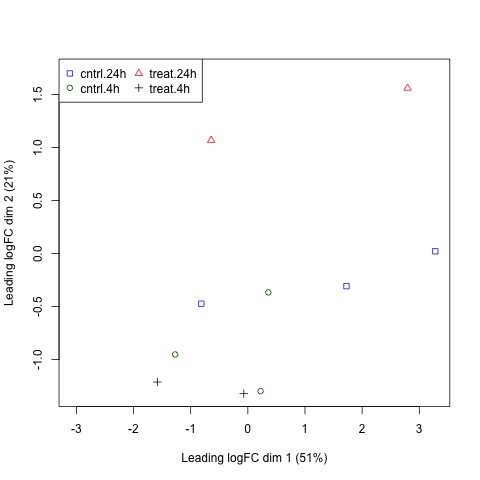
#Create plot with legend
plotMDS(list, col=colors[group], pch=points[group])
legend("topleft", legend=levels(group), pch=points, col=colors, ncol=2)
Plots
The design matrix for our data also needs to be specified before we can perform the F-tests. The experimental design is parametrized with a one-way layout and one coefficient is assigned to each group.
#The experimental design is parametrized with a one-way layout,
# where one coefficient is assigned to each group
design <- model.matrix(~ 0 + group)
colnames(design) <- levels(group)
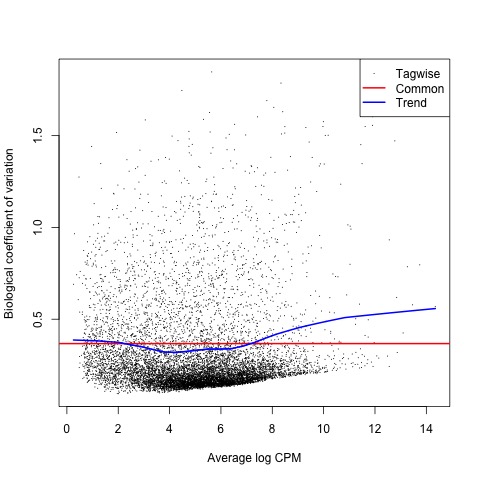
With the normalized gene counts and design matrix we can now generate the negative binomial (NB) dispersion estimates using the estimateDisp function. The NB dispersion estimates reflect the overall biological variability under the QL framework in edgeR. This allows us to use the plotBCV function to generate a genewise biological coefficient of variation (BCV) plot of dispersion estimates.
#Next, the NB dispersion is estimated
list <- estimateDisp(list, design, robust=TRUE)
#Visualize the dispersion estimates with a BCV plot
plotBCV(list)
Plot
Next, we estimate the QL dispersions for all genes using the glmQLFit function. This detects the gene-specific variability above and below the overall level. The dispersion are then plotted with the plotQLDisp function.
#Now, estimate and plot the QL dispersions
fit <- glmQLFit(list, design, robust=TRUE)
#Create plot
plotQLDisp(fit)
Plot
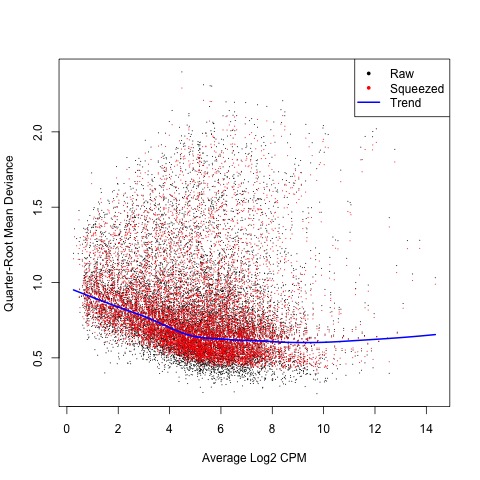
Now we are ready to begin defining and testing contrasts of our experimental design. The first comparison we will make is used to test our hypothesis that the means of the treatment factor are equal.
We can use the glmTreat function to filter out genes that have a low logFC and are therefore less significant to our analysis. In this example we will use a log2 FC cutoff of 1.2, since we see a relatively large number of DE genes from our test. The DE genes in our test are plotted using the plotMD function.
#Test whether the average across all cntrl groups is equal to the average across
#all treat groups, to examine the overall effect of treatment
con.treat_cntrl <- makeContrasts(set.treat_cntrl = (treat.4h + treat.24h)/2
- (cntrl.4h + cntrl.24h)/2,
levels=design)
#Look at genes with significant expression across all UV groups
anov.treat_cntrl <- glmTreat(fit, contrast=con.treat_cntrl, lfc=log2(1.2))
summary(decideTests(anov.treat_cntrl))
#Create MD plot of DE genes
plotMD(anov.treat_cntrl)
abline(h=c(-1, 1), col="blue")
#Generate table of DE genes
tagsTbl_treat_cntrl.filtered <- topTags(anov.treat_cntrl, n=nrow(anov.treat_cntrl$table), adjust.method="fdr")$table
write.table(tagsTbl_treat_cntrl.filtered, file="glm_treat_cntrl.csv", sep=",", row.names=TRUE)
Plot
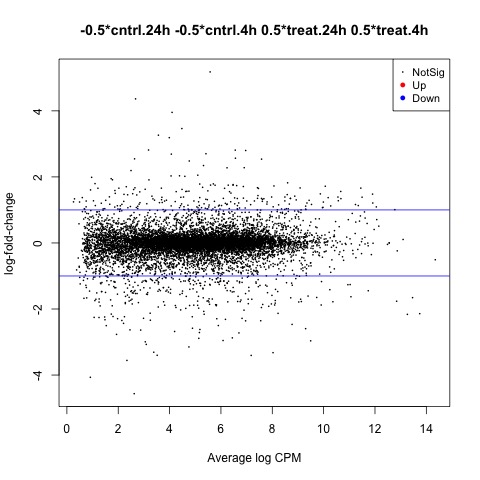
The second comparison we will make is used to test our hypothesis that the means of the hours factor are equal.
#Test whether the average across all tolerant groups is equal to the average across
#all not tolerant groups, to examine the overall effect of tolerance
con.24h_4h <- makeContrasts(set.24h_4h = (cntrl.24h + treat.24h)/2
- (cntrl.4h + treat.4h)/2,
levels=design)
#Look at genes with significant expression across all UV groups
anov.24h_4h <- glmTreat(fit, contrast=con.24h_4h, lfc=log2(1.2))
summary(decideTests(anov.24h_4h))
#Create MD plot of DE genes
plotMD(anov.24h_4h)
abline(h=c(-1, 1), col="blue")
#Generate table of DE genes
tagsTbl_24h_4h.filtered <- topTags(anov.24h_4h, n=nrow(anov.24h_4h$table), adjust.method="fdr")$table
write.table(tagsTbl_24h_4h.filtered, file="glm_24h_4h.csv", sep=",", row.names=TRUE)
Plot
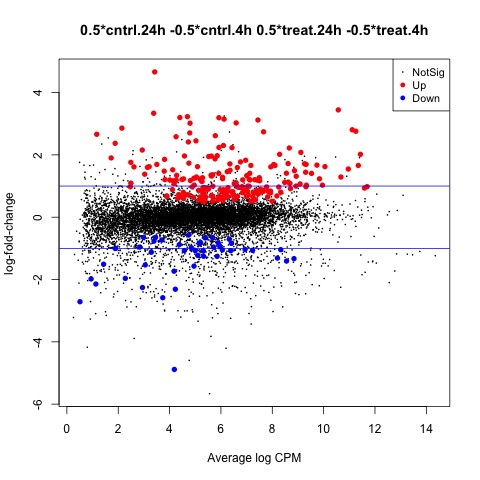
The final comparison we will make is to test our hypothesis that there is no interaction between the two factors (treatment and tolerance). To make our last contrast we will test whether the mean effects of the two factors are equal.
#Test whether there is an interaction effect
con.interaction <- makeContrasts(set.interaction = ((treat.4h + treat.24h)/2
- (cntrl.4h + cntrl.24h)/2)
- ((cntrl.24h + treat.24h)/2
- (cntrl.4h + treat.4h)/2),
levels=design)
#Look at genes with significant expression
anov.interaction <- glmTreat(fit, contrast=con.interaction, lfc=log2(1.2))
summary(decideTests(anov.interaction))
#Create MD plot of DE genes
plotMD(anov.interaction)
abline(h=c(-1, 1), col="blue")
#Generate table of DE genes
tagsTbl_inter.filtered <- topTags(anov.interaction, n=nrow(anov.interaction$table), adjust.method="fdr")$table
write.table(tagsTbl_inter.filtered, file="glm_interaction.csv", sep=",", row.names=TRUE)
Plot
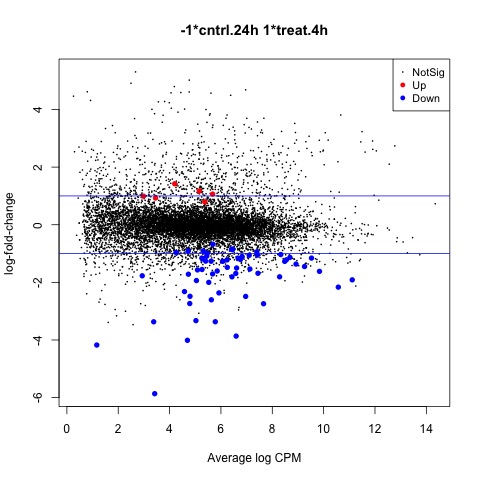
Scripts for this Lesson
Here is the R script with the code from this lesson. Also included is a R script with the plots being written to jpg files, rather than output to the screen.
Key Points
The BiocManager is is a great tool for installing Bioconductor packages in R.
Make sure to use the ? symbol to check the documentation for most R functions.
Fragments of sequences from RNA sequencing may be mapped to a reference genome and quantified.
The edgeR manual has many good examples of differential expression analysis on different data sets.
Supplemental - Introduction to Omics Data Analysis
Overview
Teaching: 0 min
Exercises: 0 minQuestions
What is bioinformatics and biostatistics?
What are the similarities between bioinformatics and biostatistics?
How can I combine R and BASH scripts to automate my data analysis workflow?
What will we be covering in this workshop?
How can I use R and BASH scripts to automate my data analysis process?
Objectives
Learn the key features of the bioinformatics and biostatistics fields.
Discover the similarities between bioinformatics and biostatistics.
Determine what coding exercises we will be performing in this workshop.
What is Biostatistics?
Biostatistics is the application of statistical methods to the designing, analyzing, and interpreting of big biological data such as genetic sequences. This requires the development and application of computational algorithms in order to conduct different complex statistical analyses.

The goal of biostatistical analyses are to answer questions and solve problems in a variety of biological fields, ranging from medicine to agriculture. For example, biostaticians are able to use mathematics to study the determining factors that impact the health of people in order to arrive at conclusions about disorders and diseases.
Checklist
Some of the common skills required of a biostatician include:
- experimental design
- collecting data
- visualizing data
- interpreting results
- computing
- problem solving
- critical thinking
- mathematical statistics
- hypothesis testing
- multivariate analysis
- analysis of correlated data
- parameter estimation
- probability theory
- modeling
What is Bioinformatics?
Bioinfotmatics is an interdisciplinary field that combines techniques and knowledge from both computer science and biology. It is a computational field that involves the analysis of complex genetic data. This commonly includes DNA, RNA, or protein sequence data.

Bioinformatics data is generated through various omics technologies used to analyze different types of biological molecules. Omics technologies include genetic, transcriptomic, proteomic, and metabolomic data.
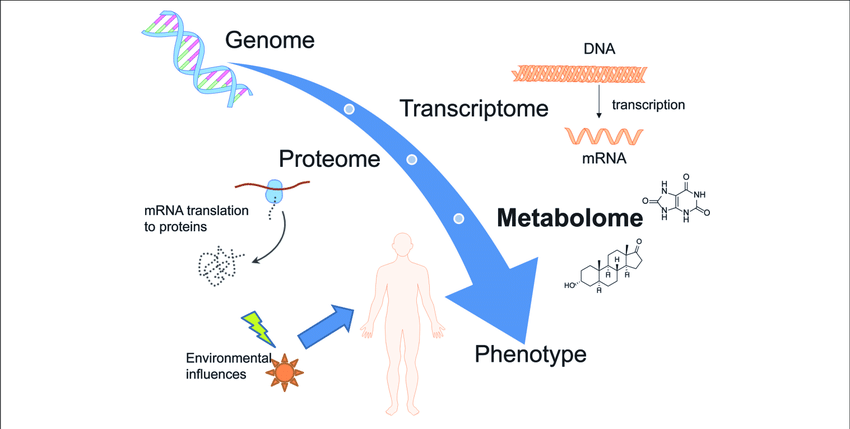
Bioinformaticians develop algorithms and use computer programming to investigate the patterns in omics data. For example, it is possible to determine the function of newly sequenced genes by comparing their protein sequences with a database of functionally annotated sequences. More powerfully, different omics data can be combined with other biological data to find complex and subtle relationships.
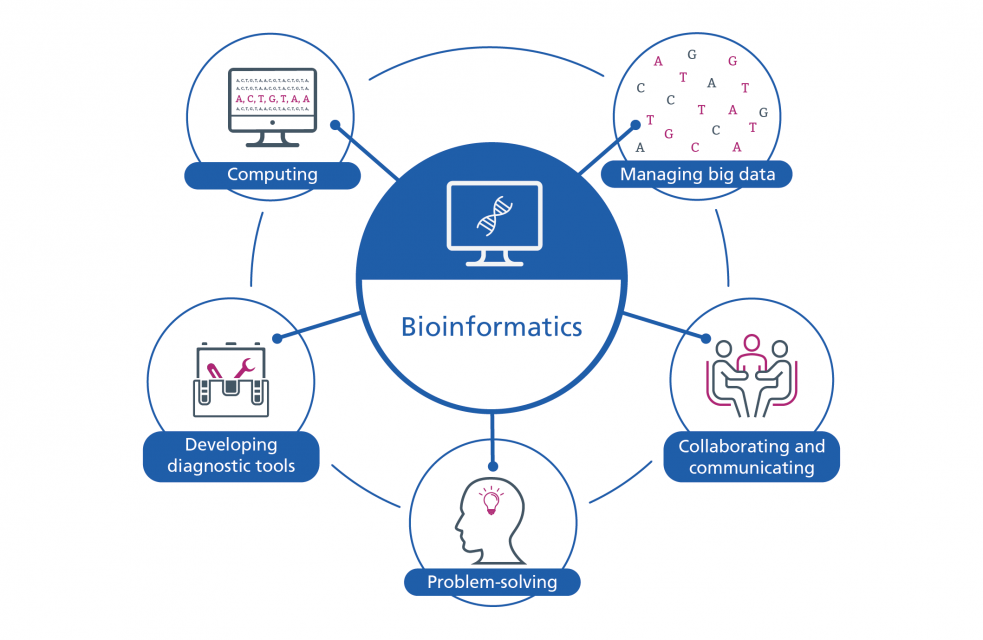
Checklist
Some of the common skills requires of bioinformaticians include:
- mining (collecting) data
- visualizing data
- computing
- problem solving
- critical thinking
- omics data analysis
- genomics
- transcriptomics
- proteomics
- metabolomics
Discussion
What are some of the similarities between the fields of bioinformatics and biostatistics?
Solution
Some of the similarities between the two fields are:
- colelcting data
- visualizing data
- computing
- problem solving
- critical thinking
Data Analysis Pipelines
Together bioinformatics and biostatistics are important interdisciplinary fields for those conducting research in a wide variety of biological sciences. A typical bioinformatics or biostatistics data analysis workflow will have multiple steps, which may include:
- experimental design
- data collection
- quality control
- data preparation
- statistical analysis of omics data
- data visualization
- interpretation of results
The following graphic depicts a general worklfow for preparing, analyzing, and visualzing results from transcriptomic RNA sequencing data.
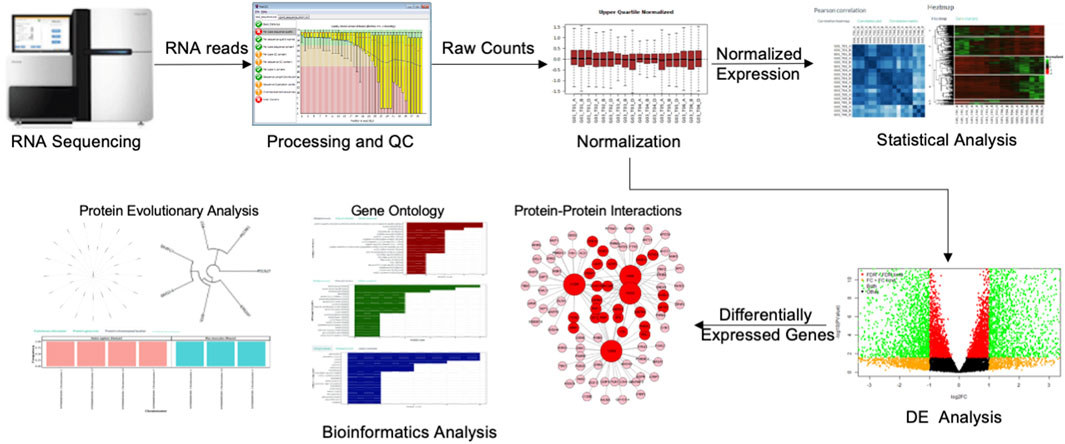
In this workshop you will gain experience in performing different aspects of bioinformatics and biostatistical data analysis by working through a scripting pipeline for analyzing gene expression data. The steps in the bioinformatics workflow for this workshop are:
- data collection - SRA toolkit
- quality control - fastqc
- data preparation
- convert genomic data format - gffread
- align transcriptomic data - hisat2
- quantify transcript alignments - featureCounts
- basic statistical analysis and visualization (exact tests) - edgeR
- advanced statistical analysis and visualization (generalized linear models) - edgeR
Pipelining & The Benefits of Scripting
The primary goal of any bioinformatics analysis workflow is to give meaning to complex biological data sets. This often requires the development of code that is generalized and can be automated to run on multiple similar data sets, which may have small differences in their structure or content.

As we have seen, the analysis of biological data using bioinformatics or biostatistical analysis is often a process that requires multiple steps. Furthermore, these steps often need to be completed in a specific order to achieve a final result. This process involves the output of a previous step being used as input to the subsequent step. This flow of outputs, to inputs, to a final result can be depicted as water flowing from one pipe to the next.

By creating a pipeline of scripts, it is possible to automate much of the bioinformatics workflow. This means making modular scripts to perform different steps of the analysis process.
BASH Scripting
We can use BASH scripting to create modular pieces of code for use in bioinformatics data analysis pipelines. BASH scripts are text files that have the .sh file extension. These are text files that you can use to save the lines of BASH code that you want the interpreter componenet of the computer operating system to execute (run).
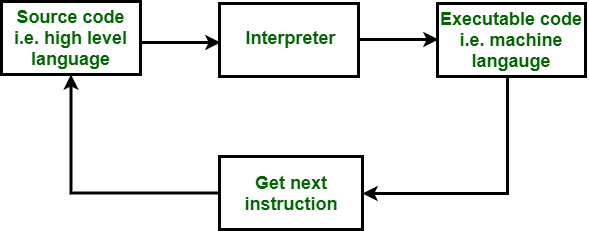
The coding exercises in this workshop are designed to give you experience with developing and running your own shell scripts using BASH programming. Additionally, you will have a chance to perform statistical analysis using your own R scripts that you run using BASH scripts. These are important skills for anyone interested in developing a bioinformatics script pipeline for analyzing large and complex biological data sets.

Key Points
Biostaticians apply statistical methods to the analysis of biological data sets.
Bioinformaticians use algorithms and computer programming to alayze omics data
There are a huge variety of omics technologies that generate data for bioinformatics analysis.
Scrpting pipelines can be used to automate complex bioinformatics analysis workflows.